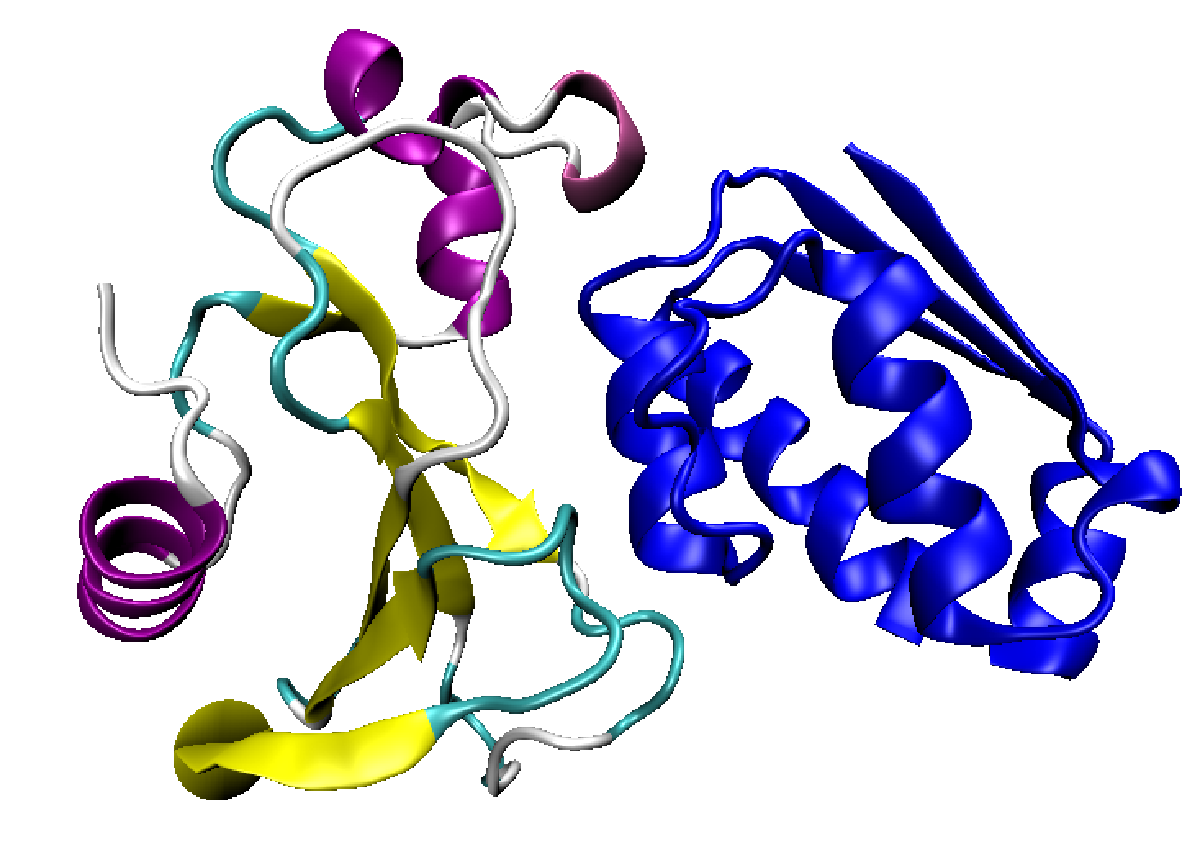

The main work horses of the cell are protein molecules. It’s the proteins that bring about development and sustain life. The instructions for building them are encoded in DNA and a large proportion of the genome is devoted to making sure the right proteins are made at the right time and at the right level.
Proteins come in a wide array of shapes and sizes that determine what jobs they do. Some need pockets to help chemical reactions proceed. Others need to form sheets or tubules. Their shape is determined by the sequence of their amino acid building blocks. Yes – another code!
I spent three years studying this particular code during my PhD at the University of York.
I used L-systems, which is a system of mathematics originally used to model plant growth, to explore how simple rewriting rules, together with modelling the protein’s environment, can be used to fold proteins to their final shape. You can read more about this work in three papers I published on the topic here, here and here.
Gemma Danks
Protein Folding with L-systems
PhD thesis, University of York, 2008Abstract#
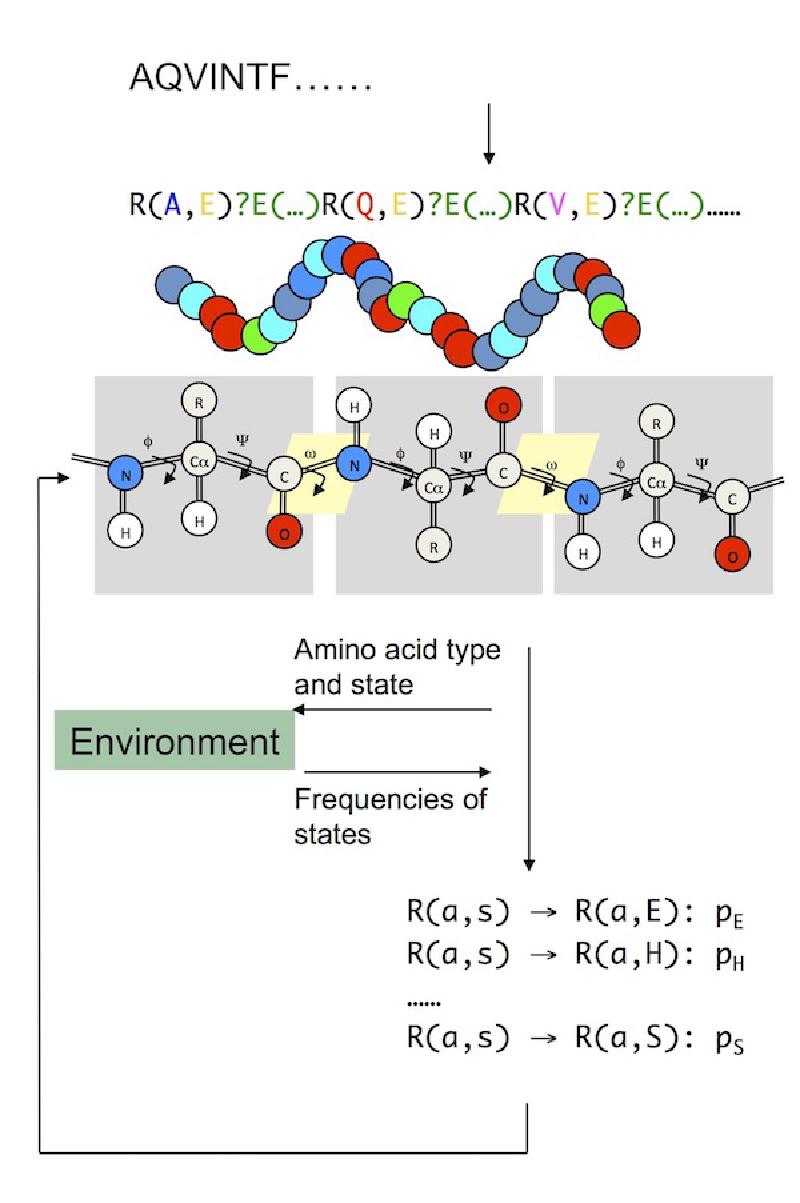
Protein folding can be viewed as an emergent phenomenon. The development of the global fold of a protein emerges due to underlying local interactions. These interactions may be modelled using a rule-based approach. L-systems are sets of parallel rewriting rules and are widely used as a mathematical framework for modelling the growth and development of plants.
This thesis presents a proof of concept of the application of L-systems to the protein folding problem. Parallel rewriting rules alter the local conformations of each amino acid residue in a polypeptide chain leading to global conformational changes. Three different L-systems models of protein folding have been developed.
A physics-based model uses parallel rewriting rules that operate on torsion angles according to local interatomic interactions. This model leads to the emergence of global conformations with protein-like compactness.
A knowledge-based stochastic model uses parallel rewriting rules that operate on the secondary structure states of residues according to probabilities that are statistically derived from native protein structures. This model leads to the emergence of global conformations with protein-like secondary structure patterns.
A third model combines physics and knowledge to give an adaptive stochastic L-systems model of protein folding. Probabilities of rewriting secondary structure states are dynamically altered at each derivation step according to local interatomic forces. This model leads to protein-like convergence to a preferred global conformation.
The physics-based, knowledge-based and combined models have been developed further to model the sequential growth of a polypeptide and the simultaneous folding of the partially formed chain. This leads to convergence to different secondary structure preferences for certain residues in the combined model. L-systems provide a natural framework for modelling cotranslational protein folding.